|
Выделение органических веществ из природных объектов
Природные органические соединения отличаются большим многообразием физических и химических свойств. Среди них встречаются относительно простые по составу жидкие вещества, обладающие значительной летучестью, твердые кристаллические соединения, сильно отличающиеся друг от друга температурой плавления и растворимостью, и, наконец, высокомолекулярные (чаще всего аморфные). Это -углеводороды, спирты, кислоты, углеводы и т. д. или их соли, эфиры и другие производные. Наиболее распространены два способа выделения органических соединений из природных продуктов: перегонка с водяным паром и экстракция растворителями. Первым способом получают сравнительно ограниченное число веществ — преимущественно те, которые входят в состав эфирных масел. Второй способ более универсален. И тем и другим способом не удается сразу же выделить индивидуальное вещество: получают лишь смесь различных соединений с близкими физическими свойствами. Разделить такую смесь на отдельные соединения иногда весьма трудно. Чтобы выделить нужное вещество и очистить его от примесей, химик-экспериментатор должен проявить много изобретательности и умения найти подходящие приемы работы. Готовых схем для этого нет. Исследуя новый продукт, приходится каждый раз искать лучшие условия получения химически чистого вещества.
Чаще всего применяются следующие методы разделения:
1. Последовательная обработка выделенной смеси различными растворителями.
2. Распределение веществ между двумя несмешивающимися растворителями. Противоточное распределение.
3. Хроматография. Электрофорез.
4. Ректификация.
5. Кристаллизация и возгонка.
6. Применение соединений включения.
Перегонка с водяным паром
Как уже упоминалось, перегонка с водяным паром имеет наибольшее значение для выделения эфирных масел. Эфиромасличные растения отличаются, как правило невысоким содержанием летучих веществ редко более 1%. Чтобы получить эфирное масло, необходимо переработать много растительного материала. Для этого нужны перегонные аппараты особой конструкции. В лабораториях пользуются обычным прибором для перегонки с водяным паром, но колбу берут возможно больших размеров и до горла загружают растением. Наклонное ее положение не обязательно (рис. 1.).
 |
 | |
Рис. 1. Прибор для отгонки эфирных масел |
Рис.2. Приемник для эфирных масел |
Приемником служит градуированная трубка с оттянутым концом. Верхний конец трубки расширен или снабжен небольшой конической воронкой (рис.2). В таком приемнике, напоминающем флорентийскую склянку, можно не только собирать эфирное масло, во и определить его выход. С помощью градуированной трубки эфирное масло легко отделяют от водного слоя. Для этого верхний конец трубки закрывают пальцем, трубку приподнимают и осторожно сливают воду, масло переносят в склянку и сохраняют для дальнейшей работы. Если нужно отогнать много масла, в качестве приемника применяют флорентийский сосуд.
Экстракция
В лабораторной практике наиболее распространена экстракция способом настаивания. Исходный материал предварительно высушивают на воздухе и измельчают. Иногда перед экстракцией сырье обрабатывают щелочами или кислотами — в зависимости от природы извлекаемых веществ и используемого растворителя. Подготовленный материал загружают в сосуд (колбу), и заливают растворителем; чаще других применяют воду, петролейный эфир, диэтиловый эфир, этиловый и изо-пролиловый спирты, хлороформ, дихлорэтан, четырехлористый углерод, бензол и иногда смеси растворителей в различных комбинациях. Настаивание продолжают несколько часов и даже суток, после чего растворитель сливают через бумажный или марлевый фильтр, а раствор обычно концентрируют. В некоторых случаях используют осадители.
Этот способ, удобный в учебных экспериментах и предварительных исследованиях, страдает существенным недостатком: он не позволяет достичь полного: извлечения. Только многократным настаиванием в новых порциях растворителя удается перевести в раствор практически все экстрактивные вещества. Когда требуется накопить извлекаемое вещество в больших количествах, применяют перколяторы из жести, оцинкованного железа или луженой меди. Им придают форму конуса или цилиндра с коническим дном. Они снабжены сливным краном и хорошо пригнанной крышкой (рис. 3).
 |
 | |
Рис. 3. Батарея перколляторов |
Рис. 4. Аппарат Сокслета |
Чтобы получить концентрированный экстракт и уменьшить расход растворителя, сосуды для настаивания или перколяторы объединяют в батареи и экстракцию ведут по принципу противотока. Сырье загружают во все сосуды (перколяторы), а растворитель заливают сначала только в один и оставляют стоять не менее суток. Затем экстракт из первого сосуда переливают во второй. Сырье после первой экстракции заливают новой порцией растворителя и опять некоторое время выдерживают. Затем раствор из второго сосуда переливают в третий, из первого снова во второй, а первый в третий раз заполняют растворителем. Так последовательно растворитель пропускают через все сосуды. В результате получается достаточно концентрированный экстракт, пригодный к дальнейшей переработке. По окончании одного цикла экстракций первый сосуд опорожняют и заполняют свежим сырьем. Первый сосуд теперь считают последним, второй — первый, третий — вторым и т. д. Чтобы ускорить экстракцию и достичь полноты извлечения при малой затрате растворителя, применяют аппарат Сокслета (рис. 4). Он весьма удобен для аналитических исследовании (например, для определения масличности семян), но в ряде случаев может быть использован и для накопления продуктов. Недостаток его — сравнительно малая емкость (обычная вместимость 20—50 г исходного материала.
Для переработки 3 — 10 кг сырья рекомендуется автоматический экстрактор (рис. 5). В его конструкции учтены особенности автоматических экстракторов, применяемых в лабораториях и в промышленности. Перколяторы между собой и с пленочным испарителем соединены системой стеклянных трубок, снабженных кранами. Достаточно перекрыть край, чтобы выключить из цикла под загрузку и разгрузку любой перколятор. По окончании процесса через патрубок 5 подают в перколятор перегретый пар, с помощью которого удается отогнать почти весь растворитель.
Пленочный испаритель, служащий для концентрирования экстракта, представляет собой конусный сосуд, соединенный с цилиндром. По впаянному в цилиндре змеевику проходит пар. Нагревается только та часть жидкости, которая соприкасается со змеевиком. Растворитель испаряется и через холодильник вновь поступает в перколятор. Очередная порция раствора омывает змеевик Сокслета тонким слоем (пленкой) и быстро вскипает. Скорость отгонки — 5—10 л в час. Уровень жидкости в испарителе остается постоянным, так как экстракт непрерывно поступает в него из перколятора через сифонную трубку. Концентрированный экстракт сливается периодически. В пожарном отношении такой испаритель безопасен. Парообразователь снабжен обратным холодильником, благодаря чему объем воды в нем во время работы не меняется.
В пленочном испарителе можно отгонять растворители из экстрактов, полученных настаиванием и в простых перколяторах. Для ускорения отгонки растворителе?, особенно высококипящих, рекомендуется создавать вакуум водоструйным насосом. К прибору в этом случае подключают более мощную охладительную систему, предотвращающую потерю растворителя.
 | | |
Pиc. 5. Схема аппаратуры для автоматической экстракции. |
|
 | | |
Рис. 6. Прибор для концентрации растворов в вакууме. | |
Концентрируют растворы и в колбах Вюрца, как и в любых других, имеющих насадку для соединения с холодильником. Если экстракта много, рекомендуется непрерывно подавать его в колбу — с той же скоростью, с какой отгоняется растворитель, В этом случае прибор собирают так, как показано на рис. 6, причем растворитель можно отгонять и в вакууме.
РАЗДЕЛЕНИЕ СМЕСЕЙ НА ИНДИВИДУАЛЬНЫЕ СОЕДИНЕНИЯ
Последовательная обработка растворителями.
Сумму веществ, полученную простым упариванием экстракта, следует подвергнуть групповому разделению и очистить от примесей. Полезно обрабатывать смесь последовательно разными растворителями. Очередность обработки зависит от природы разделяемых веществ. Если возникает необходимость освободиться от смолистых и воскообразных веществ, то в ряде случаев промывают экстракт петролейным эфиром или ацетоном. Извлечения в растворителях, не смешивающихся с водой, целесообразно разделять на нейтральную, кислую и фенольную части. Для этого разбавленный раствор экстрактивных веществ помещают в делительную воронку, туда же приливают 2 — 5-процентный раствор бикарбоната натрия и взбалтывают. После расслаивания водный раствор отделяют и повторяют операцию с новой порцией бикарбоната натрия. Содержащиеся в экстракте органические кислоты превращать в натриевые соли и переходят в водный раствор. Если прилить к раствору разбавленную серную кислоту, то соли разлагаются. Органические кислоты могут быть отделены фильтрованием или с помощью органического растворителя (эфира). Свободный от кислот экстракт затем обрабатывает 5 — 10-процентным раствором щелочи для отделения фенолов. Из освобожденного от кислот и фенолов экстракта отгоняют нацело или частично растворитель: остаются нейтральные продукты.По приведенной методике обычно исследуют растительные смолы. Чтобы выделить алкалоиды из дихлорэтанового или хлороформного экстракта, его взбалтывают с разбавленным раствором минеральной кислоты. Основания переходят в соли и растворяются в воде; остальные вещества остаются в растворителе. Кислый раствор подщелачивают, благодаря чему выделяются алкалоиды. Их отфильтровывают или извлекают подходящим растворителем (эфир, хлороформ).
Разделение с помощью несмешивающихся растворителей.
Индивидуальные вещества из смесей можно выделять, распределяя их в двух несмешивающихся растворителях. Применяются, например, такие пары растворителей: петролейный эфир — 85-процентный-спирт, бензол—водный раствор ацетона. Петролейно-эфирный или бензольный экстракт встряхивают со спиртовым или ацетоновым раствором. После расслаивания нижний слой отделяют; операцию повторяют несколько раз. Затем из каждого слоя отгоняют растворитель, а остаток кристаллизуют или хроматографируют. Надо помнить, что неполярный растворитель обогащается при этом углеводородами, простыми зфирами И подобными им веществами. Спирты, карбонильные соединения, кислоты и некоторые их производные переходят в полярный растворитель.
Следует отметить, что описанный метод позволяет осуществить только групповое разделение веществ. Тот же принцип лежит в основе противоточного распределения. Он заключается в том, что один растворитель многократно (до ста раз) промывают другим в последовательно соединенных приборах, что позволяет проводить и перемешивание слоев растворителей и разделение их одновременно во всей системе. Этот метод в отличие от предыдущего дает возможность выделять индивидуальные вещества, причем даже из таких смесей, которые другими способами неразделимы.
Хроматография
Хроматографический анализ — один из самых распространенных методов выделения и идентификации веществ. Обычно различают три основных вида хроматографии — адсорбционную, ионообменную и распределительную. В основе их лежит различная степень адсорбируемости молекул или ионов на твердом веществе (адсорбционная и ионообменная хроматография), либо различное распределение их в жидкостях, одна из которых связана с твердым носителем (распределительная хроматография).
Адсорбционная хроматография. Адсорбционная хроматография осуществляется на окиси алюминия, карбонате кальция, силикагеле, угле и других адсорбентах. Приготовление активной окиси алюминия. Если нет специальной окиси алюминия для хроматографии, 1—2 кг обычной окиси алюминия помещают в большую эмалированную кастрюлю, заливают двойным объемом 5-процентной соляной кислоты и нагревают 10 минут, все время помешивая. Если после этого жидкость не изменяет окраски бумажки конго, ее сливают, добавляют соляную кислоту и опять нагревают, добиваясь устойчивой кислой реакции в промывных водах. Обработанную таким образом окись алюминия промывают несколько раз водопроводной водой (декантировать), переносят на фарфоровую воронку к опять промывают дистиллированной водой при слабом отсасывании. Промывание прекращают, когда появится нейтральная реакция на лакмус или универсальный индикатор. После этого окись алюминия наносят тонким слоем на силикатное или органическое стекло и высушивают на воздухе. Затем переносят ее на алюминиевый или эмалированный противень и прокаливают 8—12 часов при 400—450°. Охлажденную окись алюминия ссыпают в банку с притертой пробкой или в колбу с резиновой пробкой и хранят как «кислую окись алюминия». Кислой окисью алюминия разделяют углеводороды и кётоны. Другие вещества обычно разделяют на нейтральной окиси алюминия. Для получения нейтральной окиси алюминия вводят еще одну операцию: обработанную соляной кислотой и промытую водой окись алюминия смачивают одним объемом 1-процентного раствора аммиака, затем переносят на фильтр. Промыв ее дистиллированной водой до нейтральной реакции на фенолфталеин, активируют прокаливанием, как было указано выше. Отработавшую окись алюминия можно регенерировать, для чего надо ее последовательно промыть спиртом, разбавленной соляной кислотой, водой и прокалить в описанных выше условиях.
Определение активности окиси алюминия но Брокману. В зависимости от содержания воды в окиси алюминия различают шесть степеней ее активности, с которой связана разделяющая способность адсорбента, характер и глубина вторичных процессов (окисление, дегидратация, полимеризация и др.). Окись алюминия, содержащая 2—3% влаги, имеет наивысшую активность, условно принимаемую за единицу. Активность окиси алюминия с 20% воды соответствует шести. Образцы нужной активности готовят из свежепрокаленной окиси алюминия. Для этого к ней добавляют воду и перемешивают в течение 2 часов в закрытой емкости. Сколько примерно нужно добавлять воды, можно определить по графику:

На практике приходится работать с окисью алюминия с активностью 1,5-5,0. Определение активности проводят в оттянутой с одного конца трубке длиной 12-15 см и внутренним диаметром 1,5 см. (рис. 8).

Рис. 8.
Весь суженый нижний конец заполняют ватой, накрывают вату кружком фильтровальной бумаги и насыпают испытуемую окись алюминия до высоты 5 см. Чтобы получился равномерно уплотненный слой, слабо постукивают по трубке резиновой пробкой надетой на стеклянную палочку. Затем адсорбент также накрывают кружком фильтровальной бумаги и закрепляют строго вертикально в штативе. В трубку осторожно, по палочке, наливают 10 мл раствора одного из указанных на рисунке красителей в смеси петролейного эфира и бензола (4: 1). После того как раствор почти нацело впитается, адсорбент промывают дважды смесью чистых растворителей (каждый раз берут 10 мл} и наблюдают за продвижением окрашенной зоны. Если был взят азобензол и после промывания он оказался в средней части адсорбента, то активность образца принимают за единицу. Если краситель стечет вместе с растворителем в приемник, активность равна двум и выше. Если был взят п-метоксиазобензол и полностью при промывании удержался в верхней части столбика окиси алюминия, активность также принимают за единицу. Передвижение этого красителя до средней части столбика указывает на то, что активность равна двум. Подобным образом определяют активность окиси алюминия и в остальных случаях, подбирая соответствующий краситель по данным, приведенным на рис. 8. Растворы индикаторов готовят по прописи: 40 мг красителя на 100 мл смеси петролейного эфира и бензола, взятых в соотношении 4:1. Растворители высушивают над металлическим натрием.
Предварительное хроматографирование. Для успешного разделения смесей методом хроматографии необходимо учитывать не только марку адсорбента и его активность, но и количественное соотношение разделяемой смеси и адсорбента, а также правильную последовательность использования растворителей. Применяя разные количества окиси алюминия, с помощью хроматографии можно решить различные задачи. В ряде случаев целесообразно проводить предварительное хроматографирование, т. е. групповое разделение смесей на сравнительно небольшом слое адсорбента. Ниже приводится описание типового опыта разделения смеси углеводородов и кислородсодержащих соединений (например, фракции эфирного масла). В широкую цилиндрическую или коническую делительную воронку (рис. 9) насыпают немного стеклянных или фарфоровых бусинок и поверх них кладут слой ваты. Воронку прочно закрепляют в штативе, две трети заполняют петролейным эфиром и через коническую воронку вносят окись алюминия Последнюю просеивают через небольшое сито, paзложенное над воронкой. Легким постукиванием достигается непрерывность и равномерность поступления адсорбента. Нижний конец воронки, направленый строго по оси делительной, держуат на расстоянии около 1 см от поверхности растворителя. Во время заполнения кран делительной воронки приоткрывают так, чтобы объем вытекающей жидкости соответствовал количеству внесенной окиси алюминия.
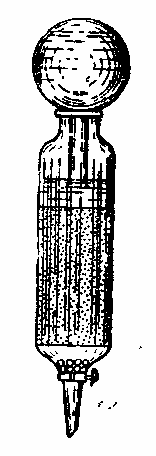 |

Рис. 10. Прибор для отгонки растворителя в вакууме. | | |
Рис. 9 |
 |
Рис.11. Капилляр |
Уровень петролейного эфира должен оставаться постоянным. Адсорбента берут в 5—15 раз больше, чем разделяемой смеси. Когда будет внесен весь адсорбент, избыток растворителя сливают так, чтобы над окисью алюминия оставался слой не более 1 мм. Затем, избегая взмучивания, покрывают окись алюминия фильтровальной бумагой. На нее наливают по стеклянной палочке 50-процентный раствор разделяемой смеси в петролейном эфире, сразу же открывают кран и начинают непрерывно сливать растворитель со скоростью 1—2 капли в секунду. Как только уровень раствора над окисью алюминия снизится до 1 мм, тем же способом добавляют немного чистого растворителя. После двукратного повторения этой операции можно быть уверенным, что вся смесь нанесена на окись алюминия, и растворитель можно налить до верха воронки. Чтобы обеспечить достаточный запас растворителя и автоматическое поступление его на адсорбент, берут узкогорлую колбу, наливают ее до верха петролейным эфиром и переворачивают горлышком вниз в делительную воронку как показано на рис. 9. Очень важно не упустить момента, когда из воронки сольется чистый растворитель (его следует возвращать в воронку) и начнет поступать первая часть раствора вещества (элюата). Этот момент можно определить, сравнивай количество поступившего и вытекшего растворителя, или заметить по появлению струек в нижней части воронки. С этого момента начинают отбор фракций равными порциями по 50, 100 мл и более, отгоняют растворитель и определяют вес фракций. Для отгонки удобно пользоваться грушевидными колбами с нормальными шлифами (рис. 10). Чтобы предотвратить бурное вскипание жидкости, вставляют в колбу длинный капилляр, перетянутый у одного конца и загнутый крючком у другого, как показано на рис. 11. Капилляр делают такой длины, чтобы загнутый конец его почти доходил до верхней пробки; за этот конец капилляр удобно вынимать из колбы, например, для регенерации. Последнюю проводят чтобы перенести фракцию из грушевидной колбы в баллончик, его шарик слегка нагревают в пламени горелки и быстро опускают тонким капилляром до дна колбочки. Жидкость втягивается почти нацело. В баллончике ее удобно взвешивать, хранить и с его помощью переливать в любой сосуд. Диаметр капилляра подбирают в зависимости от вязкости.
 |
 | |
Рис. 12. Баллончики жидкости. |
Рис. 13 |
Твердое вещество также можно количественно перенести в пробирку для перекристаллизации. С этой целью его предварительно растворяют при нагревании в минимальном количестве растворителя (в данном случае, в петролейном эфире). Перенеся раствор в другой сосуд, колбу и баллончик ополаскивают небольшим количеством чистого растворителя. Раствор ставят на кристаллизацию. Если кристаллизация не наступает, то часть растворителя отгоняют. Для этого в стаканчик с нагретой водой помещают пробирку с раствором и опускают в нее конец трубочки, соединенной с водоструйным насосом (рис. 13). Адсорбент промывают петролейным эфиром до тех пор, пока количество вещества во фракциях станет незначительным. После этого приступают к элюированию спиртом, собирая фракции, как указано выше. Описанным приемом удается грубо разделить смесь на углеводороды (петролейноэфирный элюат) и спирты, кетоны (спиртовой элюат). Исходной смеси теряется 10—15%. Если после промывания адсорбента спиртом дефицит превышает 20%, то, вероятно, в смеси находятся органические кислоты, которые могут быть отмыты водно-спиртовым раствором фосфорной кислоты. (элюаты нейтрализуют содой, концентрируют, вновь подкисляют и переводят в эфир).
Для предварительного хроматографирования применяют нейтральную окись алюминия с активностью от 2 до 3. Описанный метод иногда позволяет выделить сразу индивидуальные вещества. Однако чаще приходится объединять несколько фракций со сходными физическими свойствами и повторно хроматографировать на колонке с большим количеством окиси алюминия.
Хроматографирование на колонке. Для собственно адсорбционной хроматографии подбирают правильные размеры колонки, подходящую активность окиси алюминия и нужное соотношение адсорбента и исследуемого вещества. Колонка представляет собой трубку с краном. Отношение ее диаметра к толщине слоя адсорбента — 1:20. Свободное пространство над адсорбентом — 10—15 см. Для разделения углеводородов применяют окись алюминия с активностью 1,5—2, для разделения спиртов, азуленов, кетонов — 2—3,5, лактонов—3—4. Среднее весовое отношение углеводородов и других трудно разделяемых соединении к адсорбенту —1:80, кислородных производных — 1:40. Заполняют колонку, вносят вещество и элюируют так же, как описано выше. Только, в отличие от предварительного хроматографирования, отбирают большее количество фракций. Чем больше фракций, тем точнее разделение. Определяя вес вещества во фракциях измеренного объема и откладывая полученные значения на график, можно наглядно иллюстрировать ход процесса разделения (рис. 14). Когда один растворитель перестает вымывать заметное количество вещества, переключаются на другой. Нейтральные соединения рекомендуется последовательно элюировать петролейным эфиром, бензолом, эфиром, спиртом. Иногда применяют смешанные растворители, например петроленный эфир — бензол в соотношении 1:1.
 |
 |
 | |
Рис. 14. |
Рис. 15. Воротничковая
колбочка |
Рис. 16. Микропикнометр |
Чтобы получить вещество в чистом виде, его перекристаллизовывают или перегоняют. Перегонять удобно в воротничковых колбах (рис. 15). Желательно иметь их набор вместимостью 1, 3, 5, 10 и 20 мл. Во избежание перебрасывания вещества помещают в нижнюю часть колбы стеклянную вату. Важно, чтобы стеклянные волокна не попали в воротничок, так как иначе в него будет переходить жидкость под действием капиллярных сил. Поэтому вату сначала закладывают в стеклянную трубку, конец трубки опускают на дно и палочкой выталкивают вату. Из баллончика сливают на вату вещество (фракцию), колбу закрывают пробкой, помещают в стакан с глицерином или парафином и перегоняют в вакууме. Собравшуюся в воротничке жидкость извлекают баллончиком со слегка загнутой нижней частью капилляра. Для характеристики вещества определяют его показатель преломления и удельный вес, в оптически деятельных соединениях измеряют, кроме того, угол вращения.
Определяя показатель преломления, каплю жидкости наносят на призму рефрактометра из капилляра баллончика. Измерения проводят при 20°. Если отклонения от этой температуры невелики, то в результат вносят поправку: при повышении температуры на градус уменьшают его на 0,0004, при понижении на градус — увеличивают на столько же. Так получают приведенное значение. Удельный вес устанавливают с помощью пикнометра емкостью 0,5 и 1 мл. Если имеется всего 10—20 мг, пользуются микропикнометром, сделанным из капиллярной трубки (рис. 16). В него осторожно втягивают жидкость через оттянутый носик с помощью резиновой пипетки, натянутой на верхний конец. Как только жидкость достигнет капилляра, она заполнит его. Перед взвешиванием пипетку снимают, а носик аккуратно обтирают фильтровальной бумагой. Микропикнометр позволяет определять плотность вещества с точностью до третьего знака, что достаточно для сравнения свойств фракций. Угол вращения плоскости поляризации удобно измерять в микрокюветах длиной 2 или 5 см. Они делаются из трубочки, диаметр отверстия которой — 1—2 мм. В этом случае на определение требуется совсем немного вещества. Результаты сводят в таблицу следующей формы:

Фракции с совпадающими константами объединяют. Если резкого изменения свойств в соседних фракциях не наблюдается, проводят их повторное хроматографирование.
Ионообменная хроматография
В основе ионообменной хроматографии лежит обратимый обмен между ионами ионообменника и ионами, содержащимися в растворе. Способностью избирательно поглощать те или иные ноны обладают синтетические смолы. Их называют ионообменными смолами или ионитами. Они широко применяются для препаративного выделения различных природных соединений и для разделения сложных смесей.
Иониты—твердые, нерастворимые в воде и органических растворителях высокомолекулярные вещества с кислыми (катиониты) или основными (аниониты) группами. Катиониты способны обменивать катионы, аниониты — анионы. И те и другие—полимерные вещества с сетчатой структурой. Этим объясняется их нерастворимость и способность к набуханию. Известные в настоящее время марки ионитов различаются главным образом ионообменной емкостью. Последняя зависит от количества, природы и степени диссоциации активных групп, фиксированных на сетках макромолекулы ионита. Активными группами в катионитах могут быть — SO3H, -CH2SO3H, — СООН, — SH; в анионитах — NH2, =NH, S=N. Ионообменная способность зависит от рН среды, концентрации хроматографируемого раствора, природы поглощаемых ионов, времени контакта и от других факторов. На ионитах разделяют смеси в колонках. В качестве колонки может быть взята обычная стеклянная трубка. Один конец ее оттягивают, а на другом впаивают отвод (рис. 17).
 |
 | |
Рис. 17 |
Рис. 18 |
В хорошо пригнанную резиновую пробку вставляют небольшую изогнутую под прямым углом трубочку. На все выводы из колонки надевают резиновые шланги с зажимами. Этими шлангами колонку можно соединить с водонапорным сосудом (склянкой с тубусом внизу). Ионит, выбранный для разделения, измельчают, просеивают и, отвесив нужное количество, помещают в стакан с водой. На другой день набухшую смолу переносят в колонку. Здесь ее регенерируют и переводят в нужную для работы форму, подавая из напорного сосуда через верхнюю трубку разбавленные растворы кислот, щелочей или солей. Такой обработкой ионообменник переводят в Н-или ОН-форму или же заряжают другими ионами. Затем ионообменный фильтр тщательно промывают дистиллированной водой (до отрицательной реакции на соответствующий ион). Промывать целесообразно противотоком, присоединив напорный сосуд к нижнему выводному шлангу. Помимо того, что при этом ионит отмывается, он еще разрыхляется и освобождается от воздуха. Как во время работы, так и в перерывах ионит должен быть покрыт слоем жидкости. Когда ионообменник подготовлен, через колонку начинают фильтровать исследуемый раствор. Скорость фильтрации — не более 0,5 л в час. Так как произойдет обмен ионами, на ионите окажутся ионы веществ, предназначенных для разделения. В силу различной основности адсорбированных соединений, в слое ионита произойдет последовательный двойной обмен: вещества с большей основностью окажутся вверху, с меньшей—продвинутся вглубь ионита. Часто этот этап работы называют сорбцией. Ее завершают, как правило, промыванием ионита водой. Заключительная стадия — десорбция, т. е. вытеснение из ионообменника сорбированных ионов. Для этого вводят в колонку определенный объем раствора сильного электролита (чаще всего минеральной кислоты или щелочи), а затем промывают ее водой. Вытекающий из колонки раствор собирают по фракциям. Чистые вещества из них выделяют подходящим способом. Таким образом, многократно повторяя сорбцию и десорбцию, разделяют значительные объемы растворенных смесей на сравнительно небольших количествах смолы. Это позволяет применять иониты в промышленных масштабах для деминерализации воды, очистки гидролизатов, извлечения алкалоидов, органических кислот, редких металлов и т. д. Отечественная промышленность вырабатывает много различных ионообменных смол: катиониты КУ-1, КУ-2, СБС, СДВ-3, аниониты АН-1, АН-2Ф, ММГ-1, ПЭ-9, ДЭ-10 и др.
Распределительная хроматография
Методом распределительной хроматографии можно разделить смесь, компоненты которой имеют различные коэффициенты распределения между двумя несмешивающимися растворителями (фазами). Различают три вида распределительной хроматографии: одномерную, двухмерную и трехмерную. Первые два вида относятся к бумажной хроматографии, так как одно- и двухмерную хроматограммы получают на бумаге; третий вид относится к колоночной хроматографии, так как хроматограмму получают на колонке с сорбентом.
Хроматография на бумаге
Хроматографию на бумаге применяют для разделения микроколичеств смеси веществ. Этот метод приобрел огромное значение в исследовании белков, углеводов, жиров, антибиотиков, гормонов, каротиноидов, алкалоидов и многих других природных соединений. Вначале им пользовались главным образом для аналитической идентификации соединений. В настоящее время он применяется и для препаративного выделения чистых веществ из весьма сложных смесей. Содержащаяся в бумаге сорбированная вода служит неподвижной фазой, сама бумага — носителем. В качестве подвижной фазы применяется смесь органических жидкостей и воды в разных соотношениях. В этом методе проявляется также вполне выраженная адсорбция, поэтому он не воспроизводит распределительную хроматографию в чистом виде. Есть два варианта вертикальной хроматографии на бумаге — восходящая и нисходящая. (Известная теперь горизонтальная, или круговая, хроматография здесь не рассматривается). Чтобы получить восходящую одномерную хроматограмму, вырезают из специальной (хроматографической) бумаги полоску длиной 30—40 см, проводят черным карандашом линию на расстоянии 2,5 см вдоль короткого края и легким касанием капиллярной пипетки наносят на эту линию маленькие капли исследуемых растворов. Желательный диаметр каждого пятна — не более 5—6 мм, а расстояние между ними — 2—3 см. Бумагу просушивают так, чтоб испарился растворитель. Если нужно увеличить концентрацию раствора, операцию повторяют, причем каждую новую каплю наносят после высыхания предыдущей. Затем полоску помещают в герметически закрытый сосуд (широкий цилиндр, банку, остекленный ящик) с растворителем на дне. Ее подвешивают на стеклянных крючках в пробке или с помощью скрепленных между собой палочек, чтобы бумага не касалась стенок, а конец ниже места нанесения капель был погружен в растворитель (рис. 18). Растворитель, поднимаясь фронтом вверх по бумаге, встречает нанесенные смеси веществ. По мере продвижения растворителя продвигаются и компоненты смесей, но с различными скоростями, в силу чего каждый из них занимает свое определенное положение. Хроматография длится 15—20 часов, в течение которых растворитель успевает пропитать большую часть бумаги. Полоску вынимают из сосуда, высушивают и проявляют реагентом, дающим окрашенные соединения с анализируемыми веществами. Реагент наносят пульверизатором. Вместо этого бумагу можно погрузить в ванночку с реагентом. Там, где скопилось вещество, появится окрашенное пятно. Его надо сразу же зафиксировать, обведя простым карандашом, так как со временем оно может исчезнуть. Полученная таким образом картина пятен и называется хроматограммой. Качественный анализ хроматограмм, т. е. идентификацию разделенных веществ, проводят несколькими способами. Укажем на два из них. Наиболее простой — так называемый способ «свидетелей» —заключается в том, что на одной и той же полосе бумаги раздельно хроматографируют исследуемую смесь и набор известных (предполагаемых) веществ. Капли «свидетелей» и смеси наносят на бумагу в один ряд по линии старта. На хроматограмме визуально сопоставляют положение окрашенных пятен неизвестных компонентов с положением известных. Другой способ идентификации — определение коэффициента подвижности Rf* и сравнение полученных значений с литературными данными:
Rf = скорость движения вещества / скорость движения фронта растворителя
Практически Rf определяют так (рис. 19): на хроматограмме измеряют расстояние от линии старта вещества до центра пятна (Б), затем от линии старта до линии фронта растворителя (А) и определяют отношение отрезков. Иначе говоря, Rf = Б/А.
 |
 | |
Рис. 19 |
Рис. 20. |
Постоянство значения Rf зависит от условий хроматографии: качества бумаги, природы и чистоты растворителей, температуры, концентрации растворенного вещества и от других факторов. Этот способ менее надежен, чем способ «свидетелей». Нисходящая хроматография по сравнению с восходящей имеет то преимущество, что подвижный растворитель может стекать по каплям с края бумаги. Это позволяет осуществлять более четкое разделение медленно движущихся веществ с близкими значениями Rf. В верхней части герметически закрытого сосуда (рис. 20) закрепляют ванночку (лоточек) с растворителем; в него погружают тот край полоски, на который нанесены капли с веществом. Противоположный конец листа свисает, но не касается дна ее. На нижнем крае бумаги обычно вырезают зуб служащие точками стекания растворителя. Размер зубчиков определяют эмпирически. Например, при полоске шириной 14 см основание каждого зубчика 2,5 см. расстояние между их вершинами —1 см. Двухмерная бумажная хроматография по сравнению с одномерной более эффективна. К ней прибегают при анализе наиболее сложных смесей, например аминокислот в белковых гидролизатах. Для этого каплю исследуемого раствора наносят в угол квадратного листа бумаги и хроматографируют сначала в одном направлении, а затем, после просушивания процесс повторяют в другом растворителе, повернув хроматограмму на 90°.
Хроматография на колонке.
Если в колонку с силикагелем, пропитанным водой ввести раствор веществ А и Б в хлороформе (р слева ), а затем, промывать колонку хлороформом начнется распределение веществ между органическим растворителем и водой. Допустим, что коэффициент распределения вещества А значительно больше вещества Б. Тогда проходящий через колонку растворитель будет обогащаться веществом Б, так что через некоторое время оба вещества займут положение, изображенное на рис. 21, справа. В конечном счете вытекающие фракции будут содержать исходные вещества отдельно.
 |
 | |
Рис. 21 |
Рис. 22 |
Если коэффициенты распределения веществ в данной паре растворителей близки по значениям, то разделение смеси не будет столь четким. Наиболее подходящие растворители подбираются опытным путем. Для практической работы берут обычную хроматографическую колонку и снабжают ее большой капельной воронкой, укрепленной на пробке. Колонку заполняют специально подготовленным носителем (снликагелем, бумажным порошком и т. п.). В качестве примера приведем один из вариантов подготовки силикагеля. Определенное количество жидкого стекла (уд. в. 1,35) вдвое разбавляют водой и вводят в раствор несколько капель индикатора — метилового красного. Все-время помешивая стеклянной палочкой, осаждают из раствора 10 н. соляной кислотой гель. Кислоту прекращают приливать тогда, когда смесь в течение 30 минут не меняет светло-розовой окраски. Комочки разбивают и опять добавляют соляную кислоту до кислой реакции. На другой день сифоном сливают слой жидкости над гелем, заливают остаток водой, дают отстояться и сновa сифонируют. После этого гель отфильтровывают на воронке Бюхнера через полотно, многократно промывая дистиллированной водой до полного исчезновения кислой реакции на конго. Промытый осадок переносят в стакан, заливают водой и фракционируют взмучиванием, отделяя ту фракцию, которая оседает в первые 10 минут. Ее переносят на фильтр и, для удаления избытка воды, промывают 95-процентным спиртом. силикагель высушивают до постоянного веса сначала при 75—800, потом при 105°. Порошок освобождают от крупных частиц просеиванием через шелковое сито и сохраняют в банке с притертой пробкой. В продаже есть готовые образцы силикагелей, которые можно использовать в ряде случаев. Однако они не всегда однородны и требуют дополнительной обработки. Заполнение колонки, внесение разделяемой смеси, элюация и обработка фракцией проводятся так же, как в адсорбционной хроматографии.
Газожидкостная хроматография
В последние годы в анализе и разделении эфирных масел и других природных смесей на составные части все большее значение приобретает газожидкостная распределительная хроматография, которая широко используется для выделения индивидуальных углеводородов из природного газа, нефти и др. Она основана на многократном перераспределении смеси паров между движущимся газом и жидкостью, адсорбированной на большой поверхности инертного носителя. В качестве неподвижной жидкости применяют дибутилфталат, силиконовое масло, а также другие химически стойкие и высококипящие вещества. Носителем служит измельченный кизельгур или фарфор. В сравнении с описанной выше распределительной хроматографией данный метод отличается очень высокой эффективностью, превышающей в десятки раз эффективность лучших ректификационных колонок. Достигается это значительным увеличением длины пути, на которой происходит контакт разделяемых веществ с жидкостью и газом. Другое важное достоинство газожидкостной хроматографии — возможность вести процесс при больших скоростях газового потока в автоматическом обнаружение даже небольших количеств вещества, содержащихся в выходящем из колонки газе. Хроматографирование продолжается всего 20-30 минут. Колонка, заполненная носителем и адсорбентом, может быть использована многократно. В настоящем руководстве не ставилась цель описывать примеры разделения веществ методом газожидкостной хроматографии—это выходит за рамки поставленных нами задач.
Электрофорез на бумаге.
Вещества, в молекулах которых есть полярные группировки (—NH2, — СООН и др.), можно разделить в растворах наложением электрического поля. Такой метод называется электрофорезом. Наиболее просто электрофорез выполняется на бумаге или на гелях некоторых коллоидов (например, на агар-агаре). Его проводят в приборах, изготовленных на заводах или в самих лабораториях. Основная часть прибора — сосуды с электролитом (рис. 22). В них погружают электроды, к которым подводят постоянный ток напряжением 100 — 300 в (иногда до 1000 в). Хроматографическую бумагу, обработанную буферным раствором, закрепляют в горизонтальном положении на рамках, подставках или между стеклянными пластинками таким образом, чтобы свисающие концы ее погружались в электролит. Направление движения заряженных частиц в разделяемых веществах определяет рН электролита. Рассмотрим электрофорез аминокислот. В кислом буферном растворе кислота находится в виде катиона и, следовательно, под влиянием электрического поля будет двигаться к катоду. В щелочном буфере она превратится в анион и направится к аноду. В изоэлектрической точке аминокислота будет представлять собой биполярный ион, неподвижный в электрическом поле.
Таким образом, подбирая определенный состав электролита, разделяют смесь аминокислот на основные, кислые и нейтральные. В зависимости от размеров и форм частиц (при прочих равных условиях) скорости продвижения аминокислот к электродам будут неодинаковы. В результате на электрофореграмме сложной смеси обнаруживается несколько кислот каждого типа. После электрофореза вещества выявляют реактивами, образующими окрашенные соединения. Флуоресцирующие вещества определяют по свечению в УФ-свете. С помощью электрофореза на бумаге можно изучать состав различных природных смесей, таких как кислоты, углеводы, амины, алкалоиды и др. Этот метод приобрел большое значение в исследовании белков, антибиотиков, стероидов и вошел в практику биохимических и клинических лабораторий как важный и тонкий метод не только качественного, но и количественного анализа.
Ректификация
Ректификация на колонках — весьма эффективный метод разделения смесей. Что касается природных продуктов, то ректификация имеет наибольшее значение для получения индивидуальных соединений из эфирных масел. Вещества со сравнительно близкими температурами кипения нельзя разделить однократной перегонкой ни при обычном давлении, ни в вакууме. Если смесь двух близкокипящих веществ разделить перегонкой на три фракции и каждую из них перегнать повторно, то первая фракция обогатится низкокипящим веществом, а последняя — высококипящим. Удовлетворительных результатов разделения можно достичь только многократным повторением такой фракционной перегонки. Автоматически совершаемый процесс многократного в последовательного отделения более летучих компонентов от менее летучих называется ректификацией, а приборы, в которых это происходит, — ректификационными колонками (рис. 23).

Рис. 23 |
Главная часть прибора — вертикальная трубка, заполненная стеклянными колечками или спиралями из нихромовой проволоки. Колонку соединяют с перегонной колбой (кубом), где смесь нагревается до кипения. Верхнюю часть колонки снабжают так называемой головкой: конденсатор, трубка для термометра и кран для регулирования скорости отбора дистиллята. Если ректификацию проводят в вакууме, то перегонную колбу берут с тубусом. Вставленный в тубус капилляр для подачи воздуха обеспечивает равномерное кипение смеси. Особое приспособление, служащее для отбора фракций, позволяет менять приемник, не нарушая вакуума в системе. Колонку окружают теплоизоляционной вакуумной рубашкой или устраивают регулируемый электрообогрев, который восполняет потери тепла. Температура в рубашке на 5° ниже температуры кипения фракции. Перед пуском колонки закрывают сливной кран на головке. Колбонагревателем доводят жидкость в кубе до кипения. Для регулирования температуры колбонагреватель включают в сеть через трансформатор. Пар, проходя через колонку, частично конденсируется в ней и полностью — в конденсаторе. Конденсат стекает по насадке обратно в куб. Эту жидкость называют флегмой. Она тесно соприкасается с паром, благодаря чему пар все время обогащается более летучими компонентами. Увеличивая обогрев, добиваются интенсивного вскипания и «захлебывання» колонки так, чтобы воздух был полностью вытеснен из насадки. После этого некоторое время прибор оставляют работать «на себя» для того чтобы установилось равновесие между паром и флегмой. И уж потом отбирают фракции. Скорость поступления жидкости в приемник регулируют так, чтобы количество ее было в 20—30 раз меньше количества флегмы, стекающей с головки на насадку за одинаковый отрезок времени (флегмовое число). Это способствует лучшему разделению смеси. Жидкости, возвращающейся в куб, должно быть в 2—3 раза больше жидкости, стекающей на насадку (число орошения). Такое соотношение достигается соответствующим нагревом куба. Флегмовое число и число орошения определяют приблизительно подсчетом капель. Чем больше флегмовое число и поверхность соприкосновения жидкости с паром, тем полнее разделяется смесь. Эффективность работы колонок оценивают обычно числом «теоретических» тарелочек, которое рассчитывают по результатам перегонки смеси известного состава, например бензола с дихлорэтаном. Во время разгонки важно поддерживать постоянное давление, флегмовое число и число орошения. Фракции отбирают в зависимости от показаний термометра, так как постоянная температура — признак того, что перегоняется индивидуальное вещество. Повышение температуры свидетельствует о переходном (промежуточном) состоянии. Для характеристики собранных фракций определяют показатель преломления, удельное вращение и удельный вес. Фракции с близкими свойствами объединяют.
Кристаллизация и возгонка.
Кристаллизацию и возгонку широко применяют в работе с природными веществами. Техника очистки описана в доступных практических руководствах по органической химии, поэтому здесь не приводится. Отметим только, что возгонку вещества лучше проводить в вакууме, это позволяет избежать разложения вещества и нацело удалить следы влаги и растворителя. Весьма удобен прибор, изображенный на рис. 24.
|
 Рис. 24. Рис. 24. |
Разделение веществ методом кристаллизации с мочевиной.
Мочевина способна давать с определенными веществами соединения включения — так называемые аддукты или мочевинные комплексы. На этом и основан «мочевинный» метод. Он позволяет почти количественно разделить многие такие вещества, для которых все другие способы оказываются неэффективными или трудоемкими (углеводороды, спирты, жирные кислоты и др.). Соединения включения имеют своеобразную кристаллическую решетку. Она состоит из образованных молекулами мочевины каналов, в которых находятся органические вещества. Каналы создаются вокруг внедряемой молекулы (гостя) в момент кристаллизации. Образующиеся аддукты относятся к соединениям особого типа: в них нет ни главных, ни побочных валентностей. Вандерваальсовых сил недостаточно, чтобы связать одну молекулу вещества В с одной молекулой мочевины М. Но если одна молекула В окружена многими молекулами М, то вандерваальсовы силы складываются и дают энергию, достаточную для образования соединения:

Включенная молекула как бы окружена решеткой и удерживается в ней довольно прочно. Выделить вещество можно, лишь разрушив комплекс. Это происходит при плавлении и растворении. Рентгенографические исследования микрокристаллов показали, что структура мочевины «рыхлая», тетрагональная, в соединениях же включения она приобретает более плотную, гексагональную, упаковку, решетка которой образует каналы со спиральным ходом. На рис. 25 показана модель такой структуры с одной внедрившейся молекулой цетана C16H38. Для нее возможен единственный способ размещения — в «вытянутой» форме в просвете канала. Так как внедряемые молекулы должны умещаться в этих каналах, то их пространственная конфигурация и размер имеют решающее значение для образования аддукта. Например, самая широкая часть канала в кристаллах мочевины имеет диаметр 6 ангстрем, а самая узкая — 5 ангстрем. Углеводороды нормального строения с поперечником ~4,1 А легко образуют соединения включения. Для углеводородов с метильным ответвлением уже требуется канал диаметром 5,5 А. Более разветвленные, а также непредельные и циклические соединения аддуктов не образуют.

Рис. 25 (слева) , 26 (справа).
Рис. 26 поясняет соотношение размеров молекул и канала мочевины. Легкость образования и прочность аддуктов возрастают с увеличением длины молекулы углеводородов или их производных. Они должны содержать не менее шести углеродных атомов. Продукты присоединения мочевины получаются очень просто. Уже при смешении насыщенных растворов мочевины и исследуемых соединений, например в нагретом метаноле, выделяются гексагональные кристаллы аддукта. Образование соединений включения — равновесный процесс, поэтому избыток мочевины способствует почти полному осаждению вещества. Параллельно с этим рекомендуется проводить слепой опыт. Вещество растворяют в том же количестве растворителя, но без мочевины. Если не будет осадка, значит концентрация раствора подобрана правильно. Смеси жидких органических соединений можно обрабатывать мочевиной без растворителя. Комплексы разлагают горячей водой. Мочевина при этом растворяется, а включенные вещества выделяются в виде масла или твердого продукта. Отделить их от мочевины можно или непосредственно, или подходящим растворителем (эфиром, бензолом, четыреххлористым углеродом). Иногда отгоняют летучее соединение в вакууме или с водяным паром. Кристаллизация с мочевиной применяется в исследованиях эфирных масел, растительных восков и жиров. Этот метод предложен даже для промышленного разделения насыщенных и ненасыщенных жирных кислот.
|